Phenylketonuria (PKU) is a genetic disorder found in newborns that affects the body's ability to break down the amino acid phenylalanine. When phenylalanine accumulates in the blood, it can lead to severe intellectual disabilities and developmental delays if untreated. Early diagnosis through newborn screening programs is critical for managing PKU and preventing its harmful effects. In clinical practice, a common example of PKU in a newborn includes symptoms such as lighter skin, hair, and eye color compared to family members, due to the decreased melanin production. Elevated levels of phenylalanine detected in blood samples confirm the diagnosis. Treatment involves a strict phenylalanine-restricted diet initiated shortly after birth to ensure normal growth and neurological development.
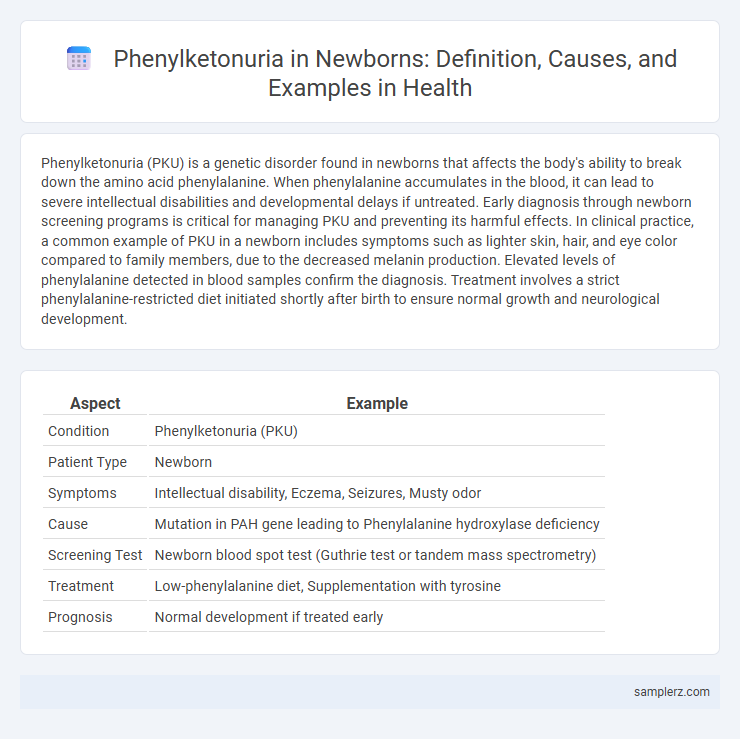
Table of Comparison
| Aspect | Example |
|---|---|
| Condition | Phenylketonuria (PKU) |
| Patient Type | Newborn |
| Symptoms | Intellectual disability, Eczema, Seizures, Musty odor |
| Cause | Mutation in PAH gene leading to Phenylalanine hydroxylase deficiency |
| Screening Test | Newborn blood spot test (Guthrie test or tandem mass spectrometry) |
| Treatment | Low-phenylalanine diet, Supplementation with tyrosine |
| Prognosis | Normal development if treated early |
Overview of Phenylketonuria in Newborns
Phenylketonuria (PKU) is a genetic metabolic disorder characterized by the inability to metabolize the amino acid phenylalanine, leading to its accumulation in the blood of affected newborns. Early detection through newborn screening programs is crucial to prevent intellectual disability and neurological damage by initiating a strict low-phenylalanine diet. Untreated PKU results in severe cognitive impairment, seizures, and behavioral problems, highlighting the importance of immediate intervention after diagnosis.
Early Signs and Symptoms in Infants
Phenylketonuria (PKU) in newborns typically presents with early signs such as a musty odor in the breath, skin, or urine due to phenylalanine accumulation. Infants may exhibit symptoms like irritability, vomiting, and delayed developmental milestones within the first few weeks of life. Early detection through newborn screening is crucial to prevent cognitive impairment and manage dietary restrictions effectively.
Causes and Genetic Factors of PKU
Phenylketonuria (PKU) is a genetic metabolic disorder caused by mutations in the PAH gene responsible for producing the enzyme phenylalanine hydroxylase, which is necessary to metabolize the amino acid phenylalanine. In newborns with PKU, the deficiency or absence of this enzyme leads to toxic accumulation of phenylalanine in the blood, resulting in brain damage if untreated. The inheritance pattern of PKU is autosomal recessive, meaning the newborn must inherit two defective copies of the PAH gene, one from each parent, to manifest the disorder.
Newborn Screening for Phenylketonuria
Newborn screening for phenylketonuria (PKU) involves a blood test typically performed within 24 to 48 hours after birth to detect elevated levels of phenylalanine, an amino acid that can cause severe intellectual disability if untreated. Early identification through this screening enables prompt dietary management to prevent cognitive impairment and neurological damage. The implementation of universal newborn screening programs has significantly reduced the incidence of PKU-related complications worldwide.
Confirming Diagnosis in Newborns
Confirming diagnosis of phenylketonuria (PKU) in newborns involves measuring blood phenylalanine levels through newborn screening tests, typically performed within 24 to 72 hours after birth. Elevated phenylalanine concentrations above 20 mg/dL indicate the need for further confirmatory tests, such as quantitative plasma amino acid analysis. Early diagnosis is critical to initiate dietary management and prevent intellectual disability and other neurological complications associated with untreated PKU.
Health Risks Associated with Untreated PKU
Untreated phenylketonuria (PKU) in newborns leads to severe neurological damage, including intellectual disability, seizures, and developmental delays, due to toxic phenylalanine buildup. High phenylalanine levels disrupt brain development, causing irreversible cognitive impairment and behavioral problems. Early diagnosis and dietary management are crucial to prevent these debilitating health risks and ensure normal growth and neurological function.
Dietary Management for Affected Newborns
Phenylketonuria (PKU) in newborns requires strict dietary management to prevent neurodevelopmental damage, primarily through a low-phenylalanine diet that limits natural protein intake. Specialized medical formulas provide essential nutrients while maintaining safe phenylalanine levels, facilitating normal growth and cognitive development. Regular blood monitoring ensures dietary adjustments are effective, minimizing risk of phenylalanine accumulation and associated complications.
Long-term Outcomes and Development
Phenylketonuria (PKU) in newborns, if untreated, leads to severe cognitive impairment, developmental delays, and neurological damage due to toxic phenylalanine accumulation. Early diagnosis through newborn screening and strict adherence to a phenylalanine-restricted diet significantly improves intellectual outcomes and reduces the risk of behavioral and psychiatric disorders. Long-term management involves regular monitoring of phenylalanine levels to support optimal brain development and prevent irreversible damage.
Real-life Case Example of PKU in a Newborn
Phenylketonuria (PKU) in newborns can be exemplified by a case where early screening detected elevated phenylalanine levels at two days old, prompting immediate dietary intervention. This early diagnosis prevented cognitive impairment, highlighting the importance of newborn metabolic screening programs. Ongoing monitoring of phenylalanine levels through blood tests ensures optimal neurological development and prevents complications associated with untreated PKU.
Support Resources for Families and Caregivers
Families and caregivers of newborns diagnosed with phenylketonuria (PKU) can access specialized support resources such as genetic counseling, dietary management programs, and local PKU foundations like the National PKU Alliance. These organizations provide comprehensive guidance on low-phenylalanine nutrition plans, medical follow-ups, and emotional support networks. Early intervention through these resources significantly enhances developmental outcomes and quality of life for PKU-affected infants.
example of phenylketonuria in newborn Infographic